Understanding the Scanning Process¶
Overview¶
Simplified overview of scanning process and preparation.
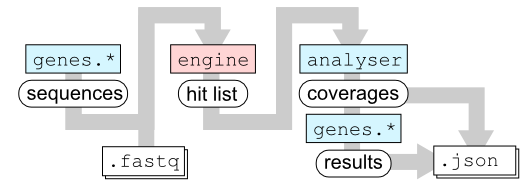
This figure illustrates the flow of data inside kvarq.
The testsuites provide a list of sequences. The C extension kvarq.engine then scans the .fastq file and finds all occurrences of these sequences. The list of these occurrences (the hit list) is then passed to the module kvarq.analyser that maps the reads onto the original sequences and creates coverages (see Coverage. This coverage is passed back to the different testsuites that calculates the final results. The coverages are saved along with the results (and optionally the hit list) into the .json file.
Testsuites 1/2¶
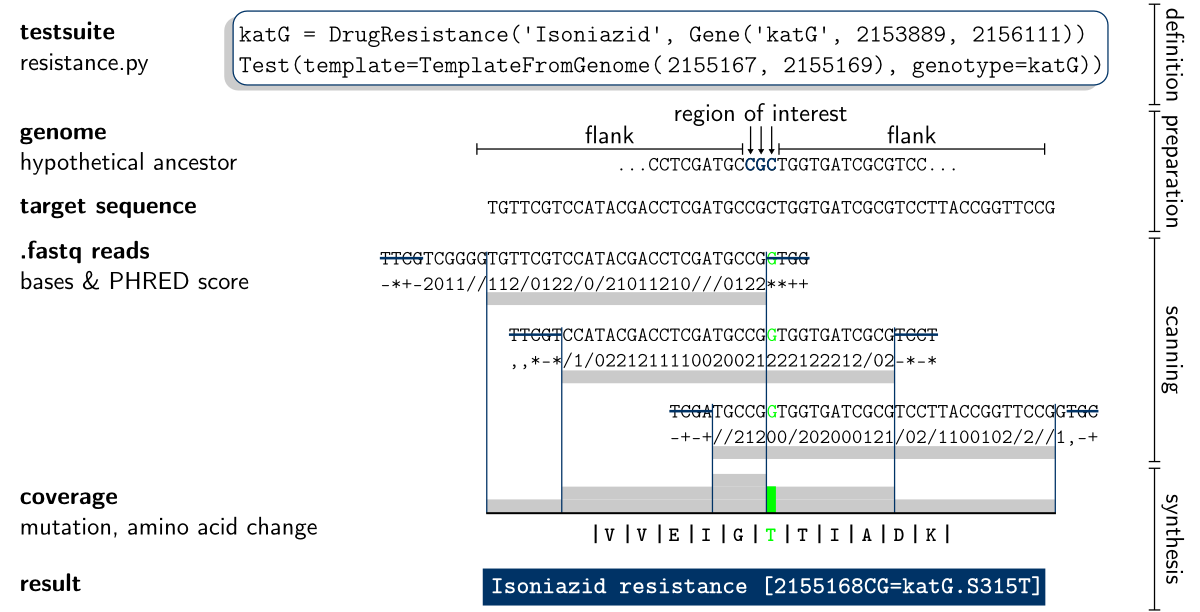
Depending on what testsuites are loaded (via the command line or the settings dialog), KvarQ performs different analysis suitable to detect different genomic markers in different organisms. The testsuite defines a template that is then used to generate a sequence which is passed along.
Engine¶
The (C extension) module kvarq.engine is the workhorse of the scanning process. It creates multiple threads that scan through the .fastq file and returns a list of kvarq.engine.Hit that describe the position and overlap of reads from the fastq with the different sequences.
This module is actually called from within kvarq.analyser and runs in a separate python thread. It provides some functions that can be called asynchronously from the main (CLI/GUI) thread to monitor the scanning process.
Analyser¶
The module kvarq.analyser takes the hit list from the kvarq.engine and applies the overlaps of the reads with the templates, creating a kvarq.analyser.Coverage object for every target sequence.
Testsuites 2/2¶
The coverages are then distributed to the different testsuites and every testsuite does some specific analysis and then reports the final results. For example, the MTBC resistance testsuite (testsuites/MTBC/resistance.py) first finds mutations and then determines whether these mutations are synonymous or non-synonymous and outputs the base mutation as well as the resulting change in amino acid if the mutation is non-synonymous.
These final results generated by the testsuites are then saved, along with the coverages, in the .json file. The file also contains all relevant scanning parameters (including testsuites and their versions).
Configuration Parameters¶
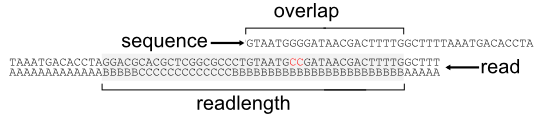
This figure illustrates the different configuration parameters for kvarq.engine
The function kvarq.engine.config() accepts the following parameters
- Amin : ASCII character of the Phred score that corresponds to the minimal quality score of a base calling to be accepted. Use method kvarq.fastq.Fastq.Q2A() to translate a Phred score into an ASCII value.
- minreadlength : After cutting the individual reads using the provided Amin, reads shorter than minreadlength are discarded.
- minoverlap : Reads that overlap (at the beginning or the end of the sequence) with less bases than the specified values are not reported.
- maxerrors : Reads that differ in more than maxerrors base positions are not considered for a match.
- nthreads : Number of threads to use in parallel for scanning the .fastq file.
These parameters can be set using command line switches or in the settings dialog.